Un aperçu de la prochaine génération d’alphafold
- Robotique
 Noesis News
Noesis News- 0
- 14 minutes de lecture
Recherche
Mise à jour des progrès: Notre dernier modèle Alphafold montre une précision considérablement améliorée et élargit la couverture au-delà des protéines à d’autres molécules biologiques, y compris les ligands
Depuis sa sortie en 2020, Alphafold a révolutionné comment les protéines et leurs interactions sont comprises. Google Deepmind et Laboratoires isomorphes ont travaillé ensemble pour construire les fondements d’un modèle d’IA plus puissant qui élargit la couverture au-delà des protéines simples à la gamme complète des molécules biologiquement pertinentes.
Aujourd’hui nous sommes Partager une mise à jour sur les progrès vers la prochaine génération d’alphafold. Notre dernier modèle peut désormais générer des prédictions pour presque toutes les molécules du Banque de données protéiques (PDB), atteignant fréquemment la précision atomique.
Il débloque une nouvelle compréhension et améliore considérablement la précision dans plusieurs classes de biomolécule clés, y compris les ligands (petites molécules), les protéines, les acides nucléiques (ADN et ARN), et ceux contenant des modifications post-traductionnelles (PTM). Ces différents types de structures et complexes sont essentiels pour comprendre les mécanismes biologiques de la cellule et ont été difficiles à prédire avec une grande précision.
Les capacités et les performances élargies du modèle peuvent aider à accélérer les percées biomédicales et à réaliser la prochaine ère de la « biologie numérique » – donnant de nouvelles informations sur le fonctionnement des voies de la maladie, de la génomique, des matériaux biorénénables, de l’immunité végétale, des cibles thérapeutiques potentielles, des mécanismes de conception de médicaments et de nouvelles plates-formes pour permettre l’ingénierie protéique et la biologie synthétique.
Série de structures prévues par rapport à la vérité au sol (blanche) de notre dernier modèle Alphafold.
Au-dessus et au-delà du pliage des protéines
Alphafold était une percée fondamentale pour la prédiction des protéines à chaîne unique. Alphafold-multitimer puis s’est étendu aux complexes avec plusieurs chaînes de protéines, suivie par Alphafold2.3, qui a amélioré les performances et élargi la couverture à des complexes plus grands.
En 2022, les prédictions de la structure d’Alphafold pour presque Toutes les protéines cataloguées connues de la science ont été rendus librement disponibles via le Base de données de structure de protéines Alphafolden partenariat avec l’Institut européen de bioinformatique d’Embl (EMBL-EBI).
À ce jour, 1,4 million d’utilisateurs dans plus de 190 pays ont accédé à la base de données Alphafold, et les scientifiques du monde vaccins contre le paludisme et avancer Découverte de médicaments contre le cancer en développement enzymes mangeurs de plastique pour lutter contre la pollution.
Ici, nous montrons des capacités remarquables d’Alphafold pour prédire des structures précises au-delà du repliement des protéines, générant des prédictions de structure très précises à travers les ligands, les protéines, les acides nucléiques et les modifications post-traductionnelles.
Performance à travers les complexes protéine-ligand (A), les protéines (B), les acides nucléiques (C) et les modifications covalentes (D).
Accélérer la découverte de médicaments
Une analyse précoce montre également que notre modèle surpasse considérablement Alphafold2.3 sur certains problèmes de prédiction de la structure des protéines qui sont pertinents pour la découverte de médicaments, comme la liaison des anticorps. De plus, la prévision avec précision des structures protéine-ligand est un outil incroyablement précieux pour la découverte de médicaments, car il peut aider les scientifiques à identifier et à concevoir de nouvelles molécules, qui pourraient devenir des médicaments.
La norme actuelle de l’industrie est d’utiliser des «méthodes d’accueil» pour déterminer les interactions entre les ligands et les protéines. Ces méthodes d’amarrage nécessitent une structure de protéine de référence rigide et une position suggérée pour que le ligand se lie.
Notre dernier modèle établit une nouvelle barre pour la prédiction de la structure protéine-ligand en surpassant les méthodes d’accueil les plus rapportées, sans nécessiter de structure protéique de référence ou l’emplacement de la poche du ligand – permettant des prédictions de protéines complètement nouvelles qui n’ont pas été caractérisées structurellement auparavant.
Il peut également modéliser conjointement les positions de tous les atomes, ce qui lui permet de représenter la flexibilité inhérente complète des protéines et des acides nucléiques lorsqu’ils interagissent avec d’autres molécules – quelque chose de non possible en utilisant des méthodes d’amarrage.
Ici, par exemple, trois cas récemment publiés et pertinents thérapeutiquement où les structures prédites de notre dernier modèle (montrées en couleur) correspondent étroitement aux structures déterminées expérimentalement (montrées en gris):
- Porcn: Une molécule anticancéreuse de stade clinique liée à sa cible, ainsi qu’une autre protéine.
- Kras: Complexe ternaire avec un ligand covalent (une colle moléculaire) d’une cible de cancer importante.
- Pi5p4kγ: Inhibiteur allostérique sélectif d’une kinase lipidique, avec de multiples implications de la maladie, y compris le cancer et les troubles immunologiques.
Prédictions pour PORCN (1), KRAS (2) et PI5P4Kγ (3).
Les laboratoires isomorphes applique ce modèle Alphafold de nouvelle génération à la conception thérapeutique des médicaments, contribuant à caractériser rapidement et avec précision de nombreux types de structures macromoléculaires importantes pour traiter les maladies.
Nouvelle compréhension de la biologie
En déverrouillant la modélisation des structures protéiques et ligands avec les acides nucléiques et ceux contenant des modifications post-traductionnelles, notre modèle fournit un outil plus rapide et précis pour examiner la biologie fondamentale.
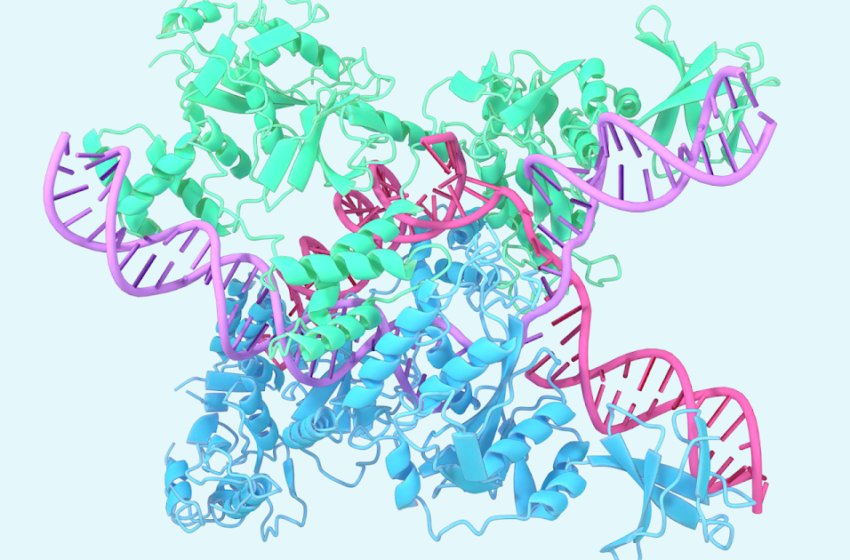
Un exemple implique la structure de Caslambda lié au CRRNA et à l’ADNpartie du Famille Crispr. Caslambda partage la capacité d’édition du génome du Système CRISPR-CAS9communément appelé «ciseaux génétiques», que les chercheurs peuvent utiliser pour changer l’ADN des animaux, des plantes et des micro-organismes. La taille plus petite de Caslambda peut permettre une utilisation plus efficace dans l’édition du génome.
Structure prédite de Caslambda (CAS12L) liée au CRRNA et à l’ADN, une partie du sous-système CRISPR.
La dernière version de la capacité d’Alphafold à modéliser ces systèmes complexes nous montre que l’IA peut nous aider à mieux comprendre ces types de mécanismes et accélérer leur utilisation pour les applications thérapeutiques. Plus d’exemples sont Disponible dans notre mise à jour de progrès.
Exploration scientifique progressive
Le saut dramatique de notre modèle dans les performances montre le potentiel de l’IA pour améliorer considérablement la compréhension scientifique des machines moléculaires qui composent le corps humain – et le monde plus large de la nature.
Alphafold a déjà catalysé des avancées scientifiques majeures dans le monde. Maintenant, la prochaine génération d’alphafold a le potentiel de faire progresser l’exploration scientifique à vitesse numérique.
Nos équipes dédiées sur Google Deepmind et les laboratoires isomorphes ont fait de grands progrès sur ce travail critique et nous sommes impatients de partager nos progrès continus.