Réutilisation des modèles de repliement des protéines pour la génération avec diffusion latente – Le blog de recherche de Berkeley Artificial Intelligence
- Intelligence Artificielle
 Noesis News
Noesis News- 0
- 18 minutes de lecture











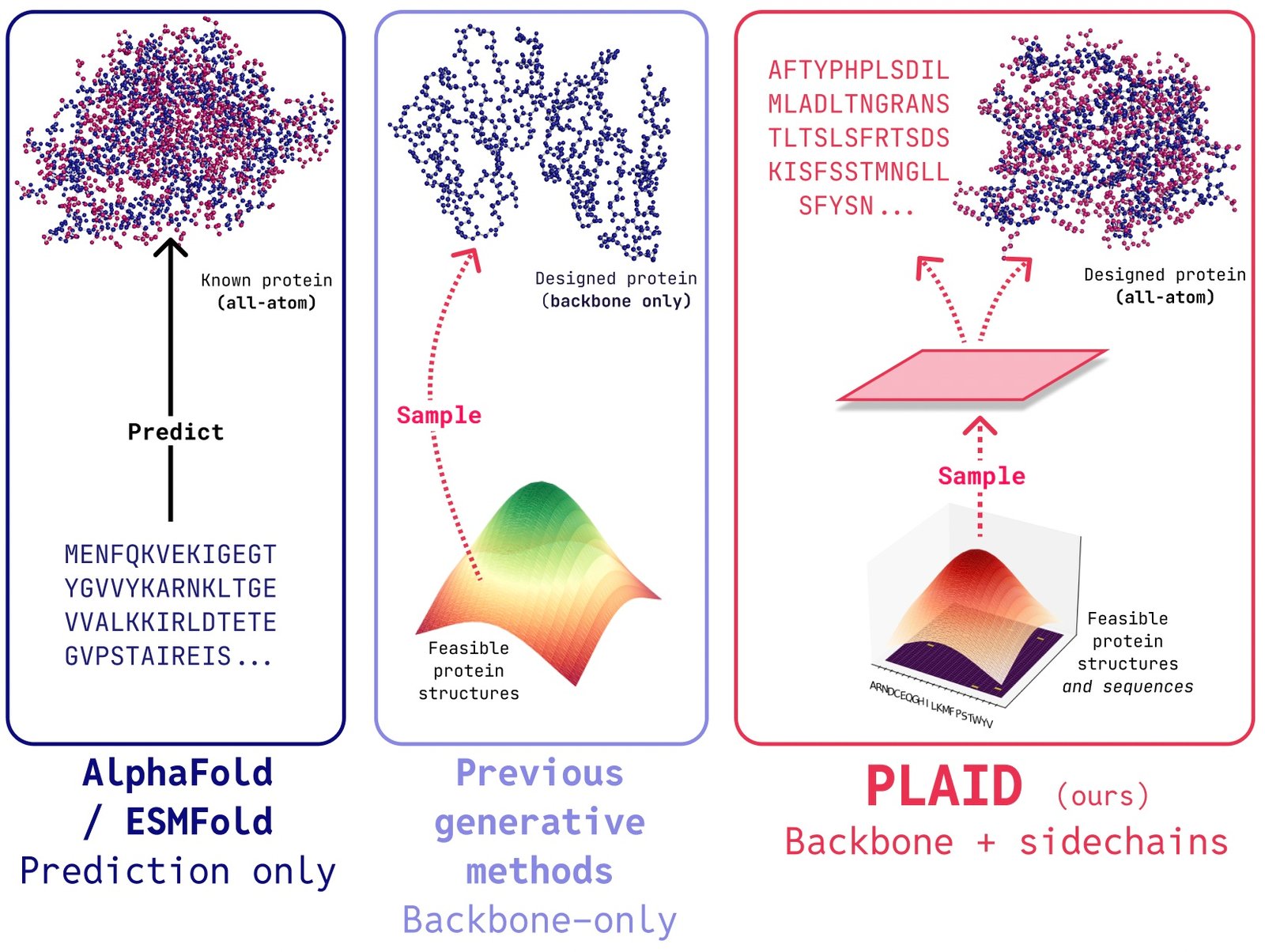
PLAID est un modèle génératif multimodal qui génère simultanément la séquence de protéine 1D et la structure 3D, en apprenant l’espace latent des modèles de repliement des protéines.
L’attribution du 2024 Prix Nobel À Alphafold2 marque un moment de reconnaissance important pour le rôle de l’IA dans la biologie. Qu’est-ce qui vient ensuite après le pliage des protéines?
Dans PLAIDnous développons une méthode qui apprend à échantillonner à partir de l’espace latent des modèles de repliement des protéines pour générer nouvelles protéines. Il peut accepter Fonction de composition et invites d’organismeet peut être formé sur les bases de données de séquencequi sont 2 à 4 ordres de grandeur plus grands que les bases de données de structure. Contrairement à de nombreux modèles génératifs de structure protéique précédents, Plaid traite du réglage du problème de la co-génération multimodale: générant simultanément à la fois une séquence discrète et des coordonnées structurelles tout-atomes continues.
De la prédiction de la structure à la conception de médicaments réels
Bien que les travaux récents soient prometteurs de la capacité des modèles de diffusion à générer des protéines, il existe toujours des limites des modèles précédents qui les rendent peu pratiques pour les applications du monde réel, telles que:
- Génération de tout atome: De nombreux modèles génératifs existants ne produisent que les atomes du squelette. Pour produire la structure de tous les atomes et placer les atomes de laasque, nous devons connaître la séquence. Cela crée un problème de génération multimodale qui nécessite une génération simultanée de modalités discrètes et continues.
- Spécificité de l’organisme: Les protéines biologiques destinées à l’usage doivent être humanisépour éviter d’être détruit par le système immunitaire humain.
- Spécification de contrôle: La découverte de médicaments et la mettre entre les mains des patients est un processus complexe. Comment pouvons-nous spécifier ces contraintes complexes? Par exemple, même après que la biologie est abordée, vous pouvez décider que les comprimés sont plus faciles à transporter que les flacons, ajoutant une nouvelle contrainte de soluabilité.
Générer des protéines «utiles»
La génération de protéines n’est pas aussi utile que contrôle la génération pour obtenir utile protéines. À quoi pourrait ressembler une interface pour ce type?

Pour l’inspiration, considérons comment nous contrôlerons la génération d’images via des invites textuelles de composition (exemple de Liu et al., 2022).
Dans Plaid, nous reflétons cette interface pour spécification de contrôle. L’objectif ultime est de contrôler entièrement la génération via une interface textuelle, mais nous considérons ici les contraintes de composition pour deux axes comme preuve de concept: fonction et organisme:
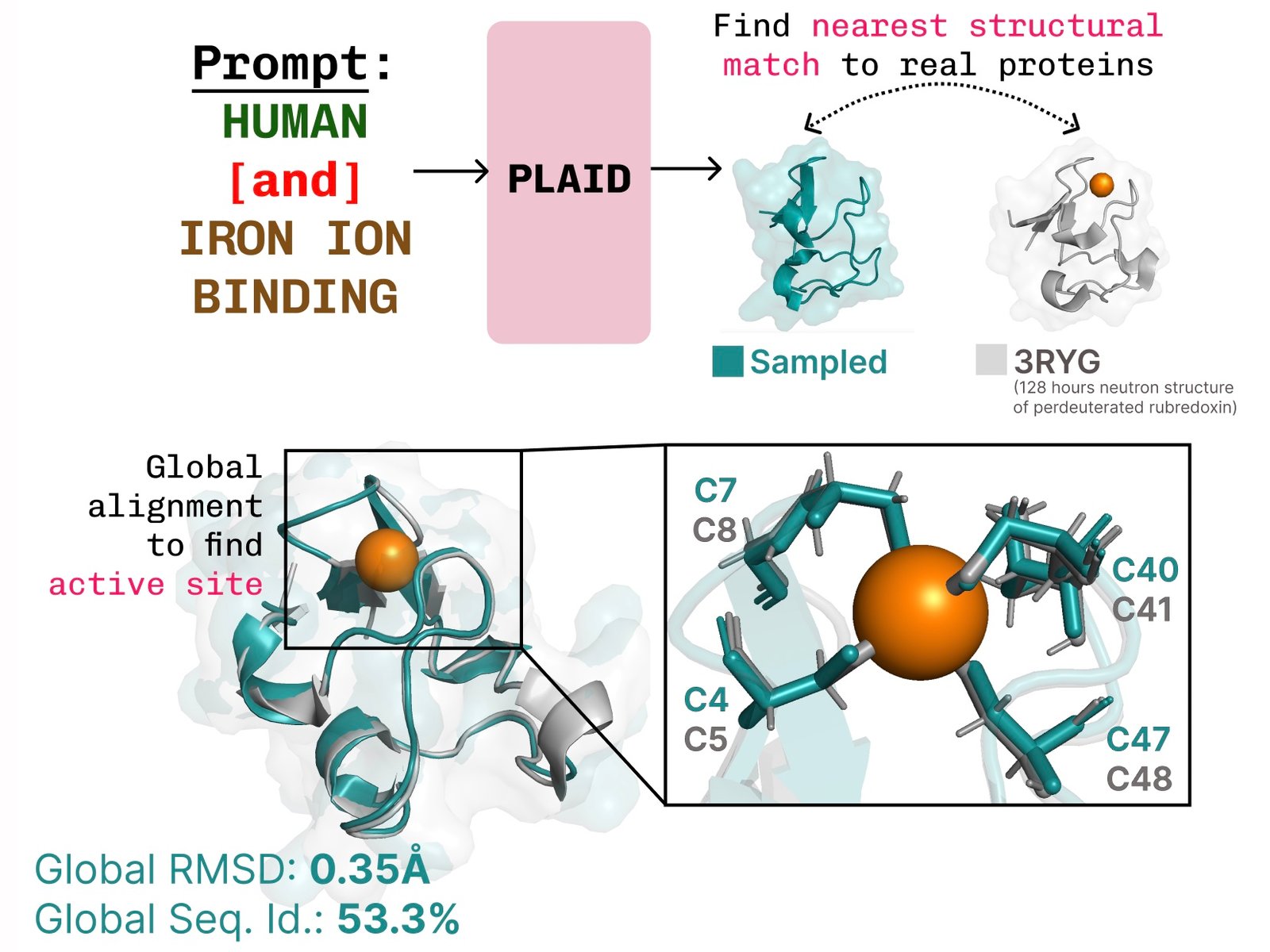
Apprentissage de la connexion fonction-structure-séquence. Plaid apprend la cystéine tétraédrique2+/ Fe3+ Modèle de coordination souvent trouvé dans les métalloprotéines, tout en maintenant une diversité élevée au niveau des séquences.
Formation en utilisant des données de formation sur séquence uniquement
Un autre aspect important du modèle Plaid est que nous avons besoin de séquences uniquement pour former le modèle génératif! Les modèles génératifs apprennent la distribution des données définie par ses données de formation, et les bases de données de séquence sont considérablement plus grandes que les bases structurelles, car les séquences sont beaucoup moins chères que la structure expérimentale.
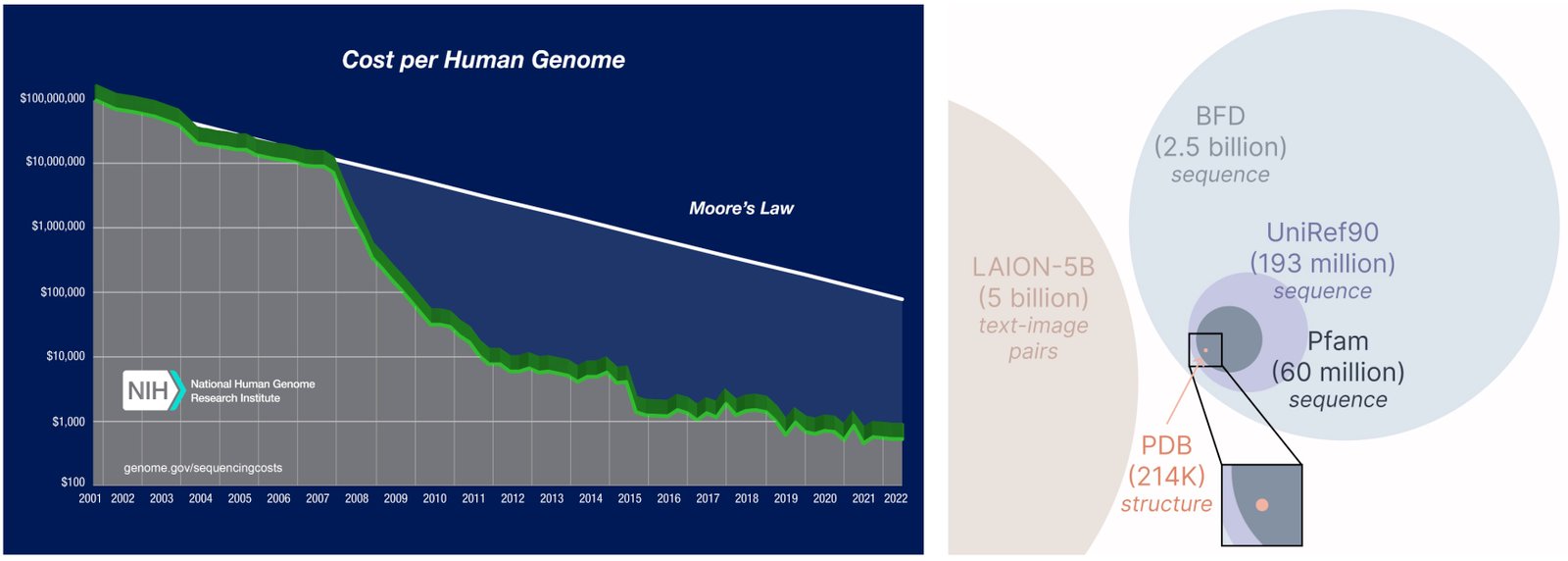
Apprendre d’une base de données plus large et plus large. Le coût de l’obtention de séquences de protéines est beaucoup plus faible que la structure de caractérisation expérimentale, et les bases de données de séquence sont 2-4 ordres de grandeur plus grands que les bases structurelles.
Comment ça marche?
La raison pour laquelle nous sommes en mesure de former le modèle génératif pour générer une structure uniquement en utilisant des données de séquence est en apprenant un modèle de diffusion sur le Espace latent d’un modèle de pliage protéique. Ensuite, pendant l’inférence, après échantillonnage de cet espace latent de protéines valides, nous pouvons prendre poids gelés du modèle de repliement des protéines à la structure du décodage. Ici, nous utilisons ESMBLOPun successeur du modèle Alphafold2 qui remplace une étape de récupération par un modèle de langue protéique.
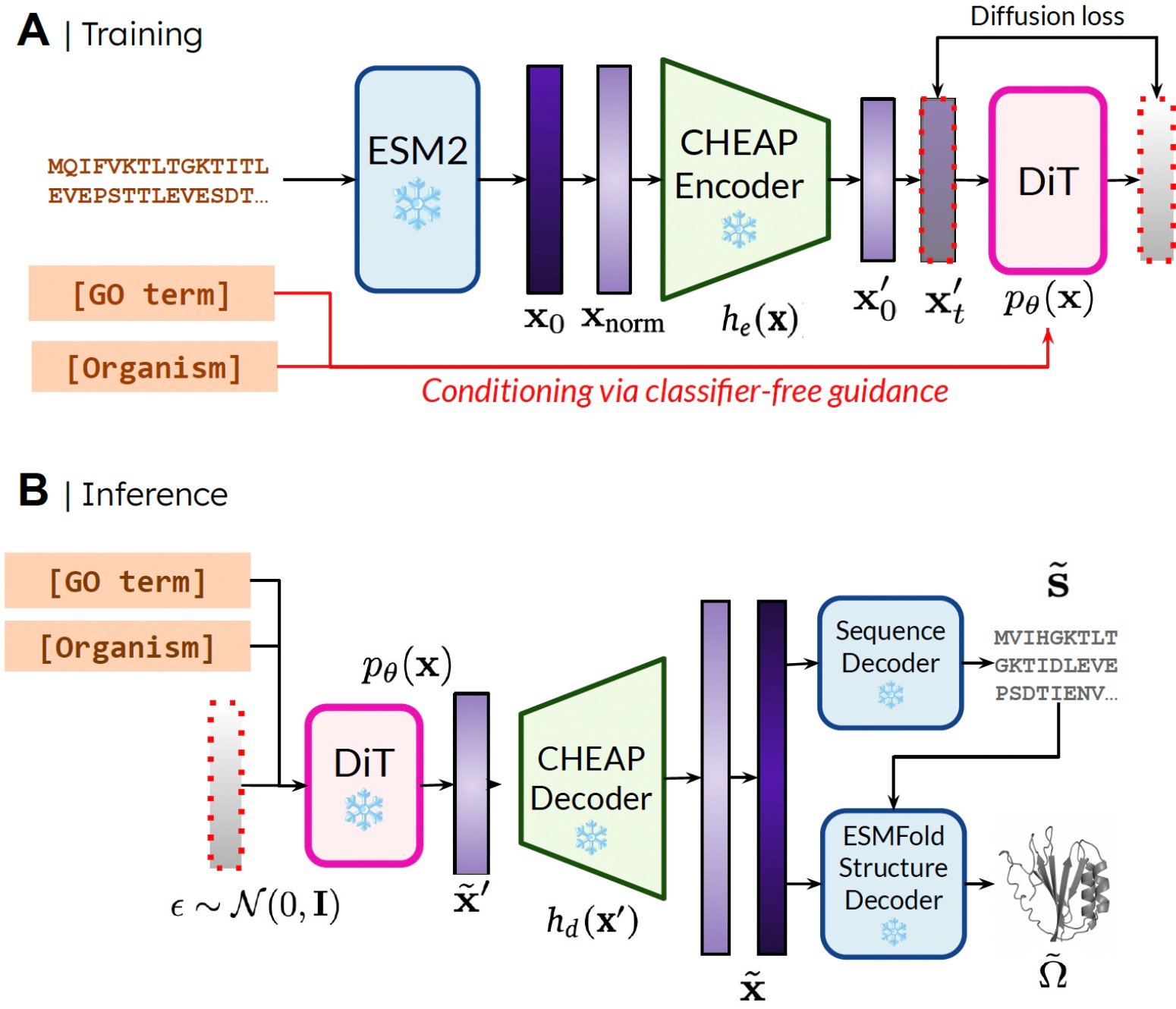
Notre méthode. Pendant la formation, seules des séquences sont nécessaires pour obtenir l’incorporation; Pendant l’inférence, nous pouvons décoder la séquence et la structure de l’intégration échantillonnée. ❄️ désigne des poids congelés.
De cette façon, nous pouvons utiliser des informations de compréhension structurelle dans le poids des modèles de repliement des protéines pré-entraînés pour la tâche de conception des protéines. Ceci est analogue à la façon dont les modèles de vision-action-action (VLA) en robotique utilisent des prieurs contenus dans les modèles de vision (VLMS) formés sur des données à l’échelle d’Internet pour fournir la perception et le raisonnement et la compréhension des informations.
Comprimer l’espace latent des modèles de repliement des protéines
Une petite ride à appliquer directement cette méthode est que l’espace latent de ESMfold – en effet, l’espace latent de nombreux modèles basés sur les transformateurs – nécessite beaucoup de régularisation. Cet espace est également très grand, donc l’apprentissage de cette incorporation finit par cartographier la synthèse d’image à haute résolution.
Pour y remédier, nous proposons également BON MARCHÉ (Sablier comprimé Adaptations de protéines)où nous apprenons un modèle de compression pour l’intégration articulaire de la séquence et de la structure des protéines.
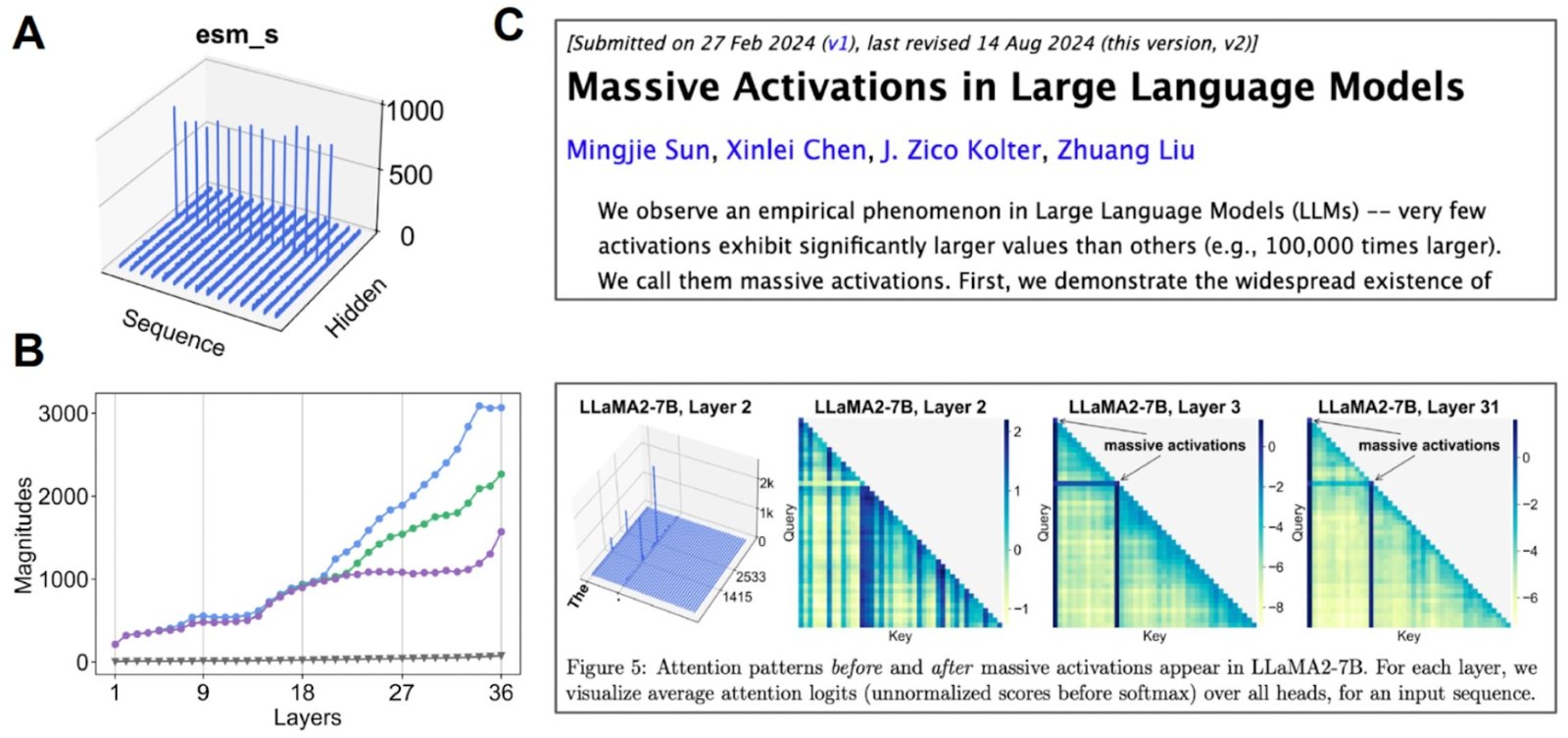
Enquêter sur l’espace latent. (A) Lorsque nous visualisons la valeur moyenne pour chaque canal, certains canaux présentent des «activations massives». (B) Si nous commençons à examiner les 3 meilleures activations par rapport à la valeur médiane (gris), nous constatons que cela se produit sur de nombreuses couches. (C) Des activations massives ont également été observées pour d’autres modèles basés sur les transformateurs.
Nous constatons que cet espace latent est en fait très compressible. En faisant un peu d’interprétabilité mécaniste pour mieux comprendre le modèle de base avec lequel nous travaillons, nous avons pu créer un modèle génératif de protéines tout atome.
Quelle est la prochaine étape?
Bien que nous examinions le cas de la séquence des protéines et de la génération de structure dans ce travail, nous pouvons adapter cette méthode pour effectuer une génération multimodale pour toutes les modalités où il existe un prédicteur d’une modalité plus abondante à une modalité moins abondante. Comme les prédicteurs de séquence à structure pour les protéines commencent à s’attaquer aux systèmes de plus en plus complexes (par exemple, Alphafold3 est également capable de prédire les protéines en complexe avec des acides nucléiques et des ligands moléculaires), il est facile d’imaginer effectuer une génération multimodale sur des systèmes plus complexes en utilisant la même méthode. Si vous êtes intéressé à collaborer pour étendre notre méthode ou pour tester notre méthode dans le laboratoire humide, veuillez contacter!
Autres liens
Si vous avez trouvé nos articles utiles dans vos recherches, veuillez envisager d’utiliser le bibtex suivant pour plaid et bon marché:
@article{lu2024generating,
title={Generating All-Atom Protein Structure from Sequence-Only Training Data},
author={Lu, Amy X and Yan, Wilson and Robinson, Sarah A and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Bonneau, Richard and Abbeel, Pieter and Frey, Nathan},
journal={bioRxiv},
pages={2024--12},
year={2024},
publisher={Cold Spring Harbor Laboratory}
}
@article{lu2024tokenized,
title={Tokenized and Continuous Embedding Compressions of Protein Sequence and Structure},
author={Lu, Amy X and Yan, Wilson and Yang, Kevin K and Gligorijevic, Vladimir and Cho, Kyunghyun and Abbeel, Pieter and Bonneau, Richard and Frey, Nathan},
journal={bioRxiv},
pages={2024--08},
year={2024},
publisher={Cold Spring Harbor Laboratory}
}
Vous pouvez également vérifier nos préimpressions (PLAID, BON MARCHÉ) et les bases de code (PLAID, BON MARCHÉ).
Une génération de protéines bonus amusante!
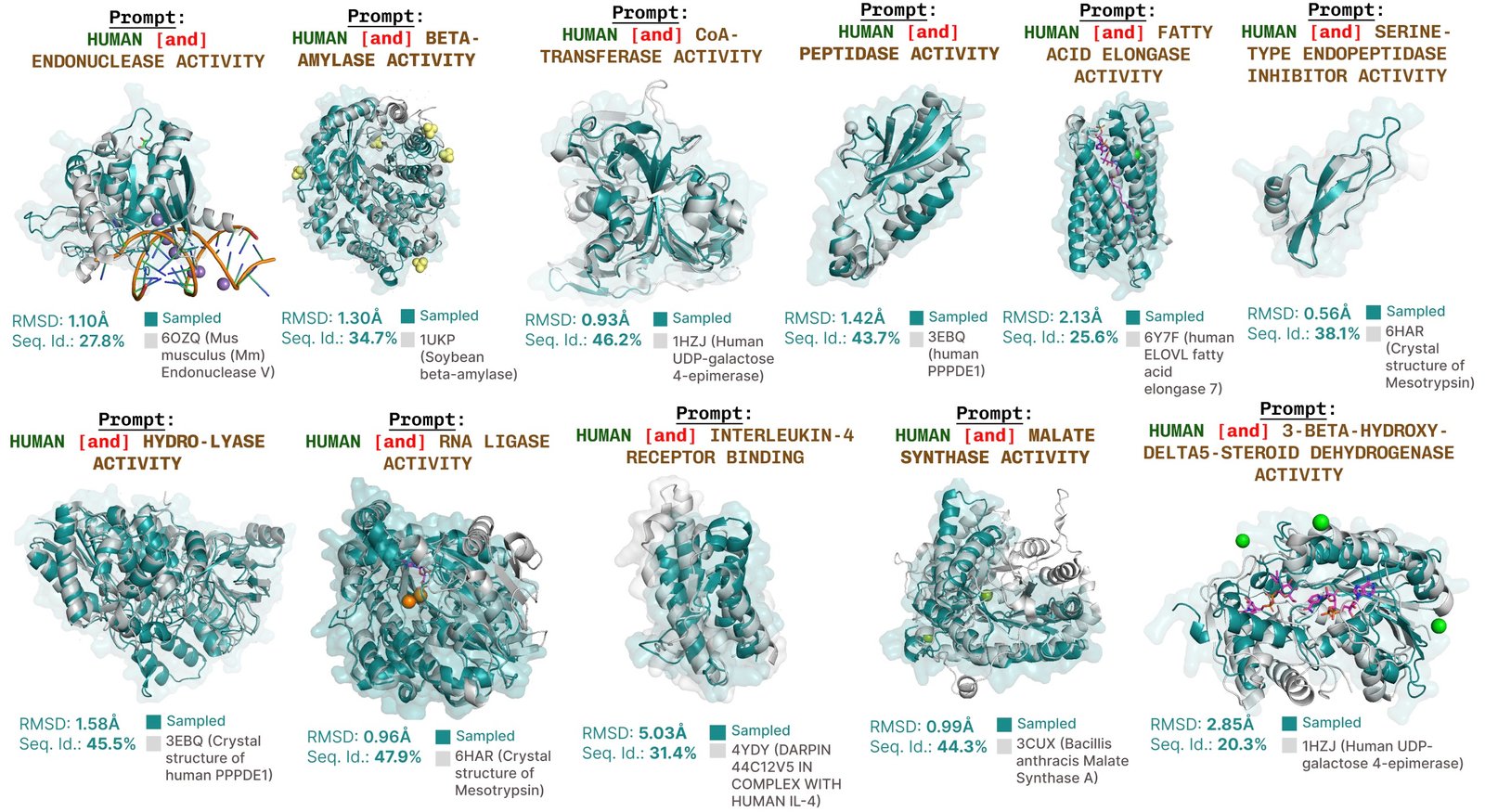
Générations supplémentaires protisées par la fonction avec plaid.
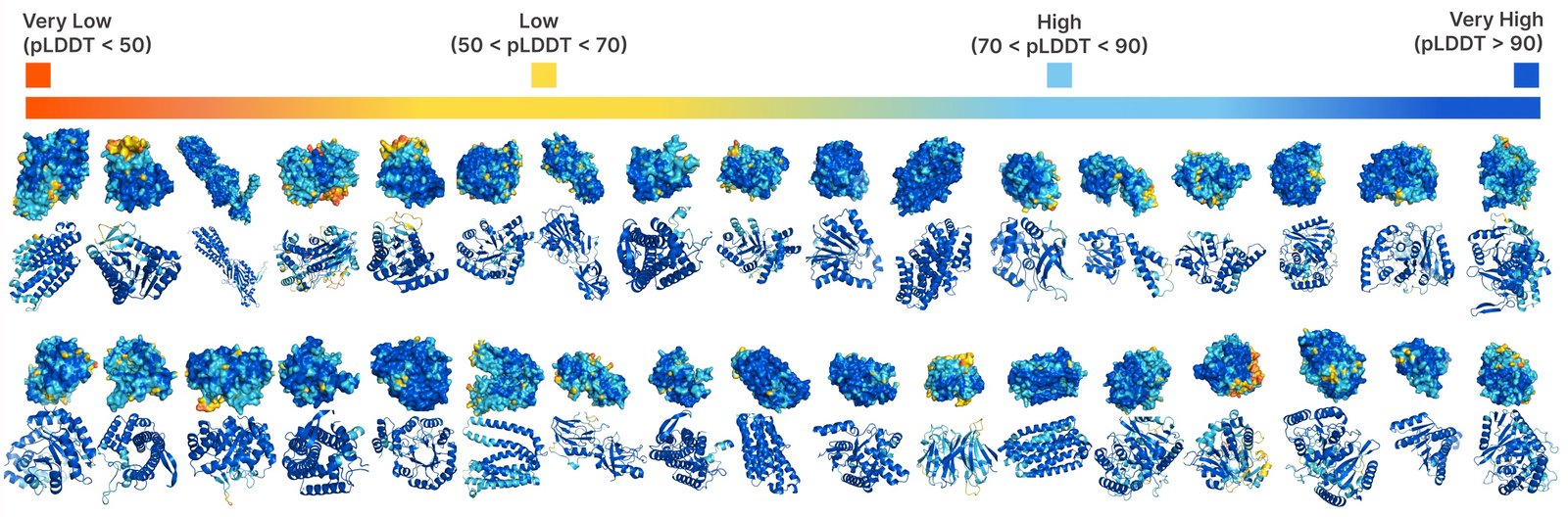
Génération inconditionnelle avec plaid.
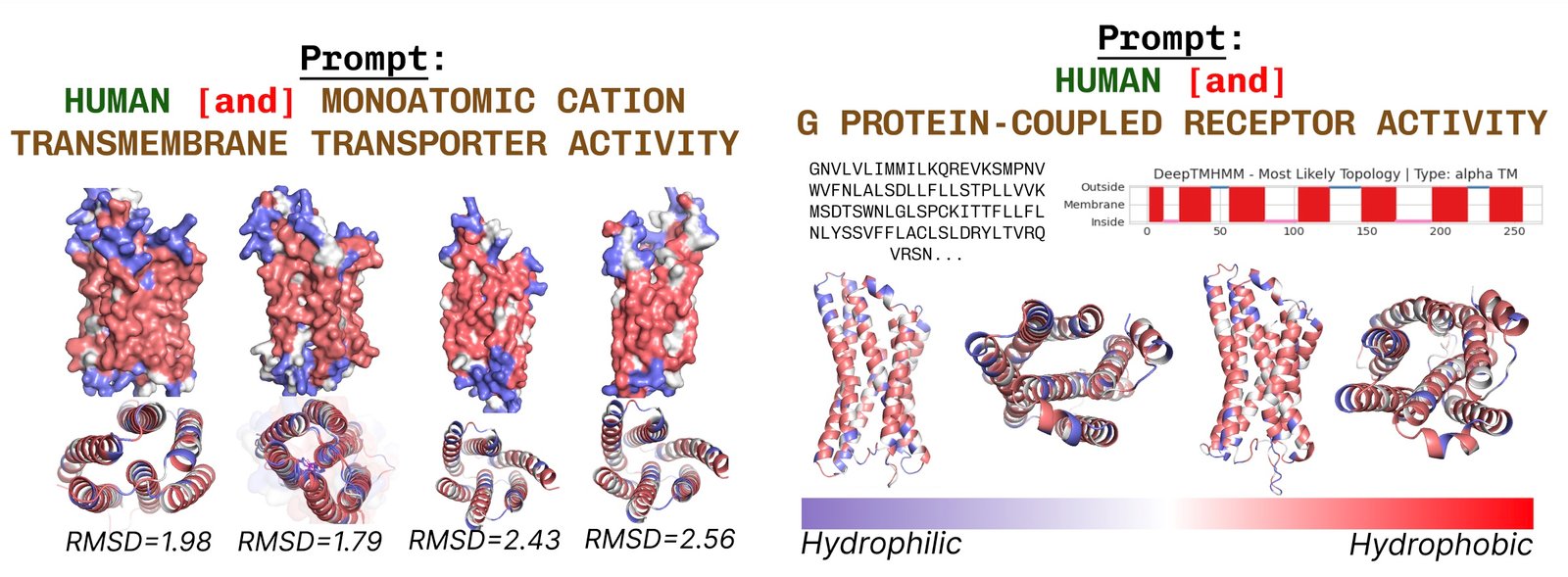
Les protéines transmembranaires ont des résidus hydrophobes au cœur, où il est intégré dans la couche d’acide gras. Ceux-ci sont constamment observés lors de l’incitation à plaid avec des mots clés de protéines transmembranaires.
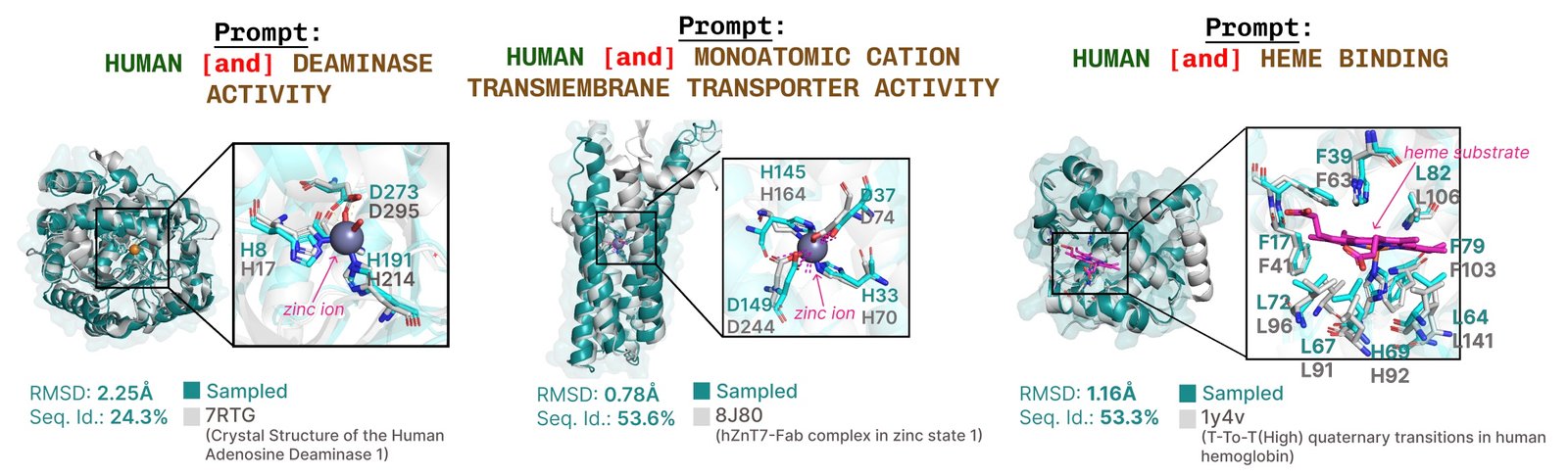
Exemples supplémentaires de récapitulation active du site basé sur la fonction de mot-clé de fonction.
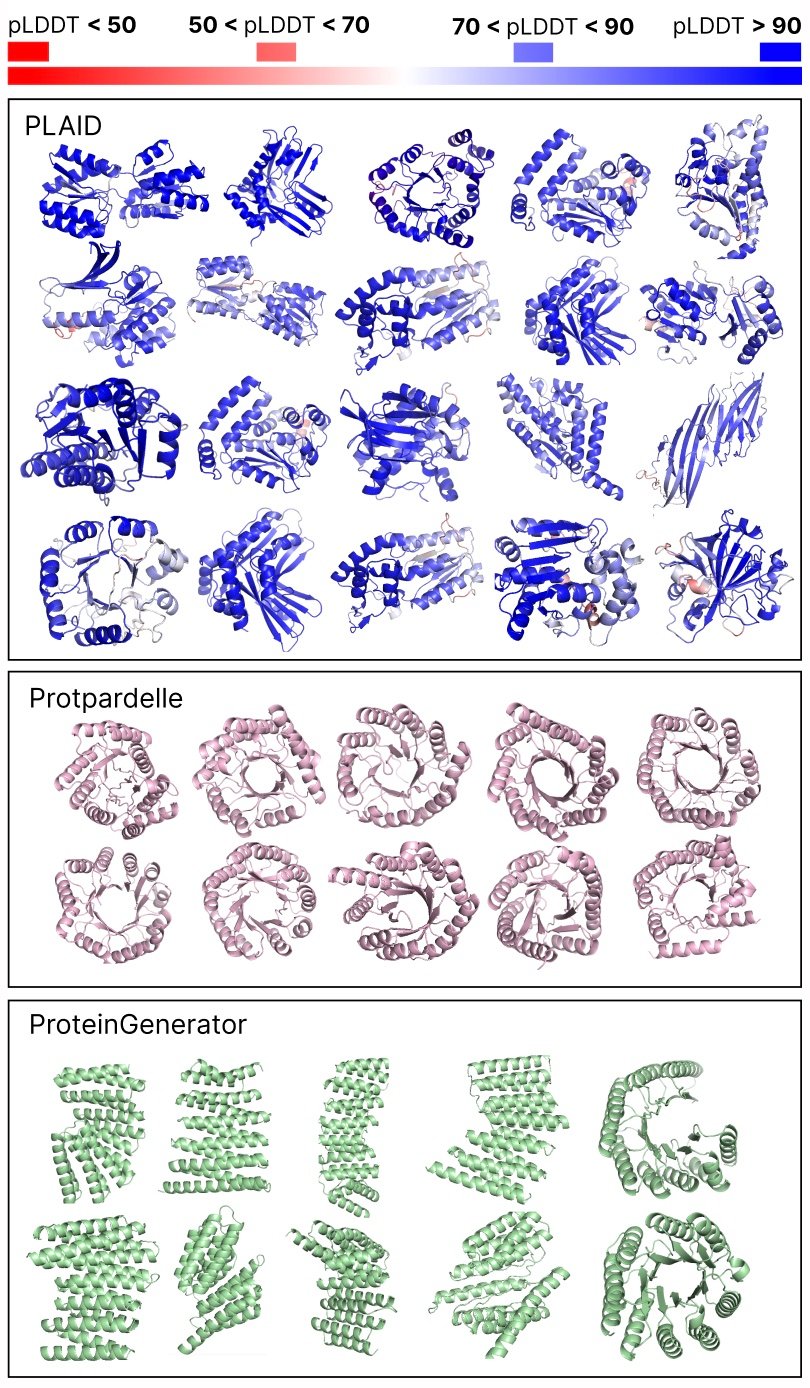
Comparaison des échantillons entre les lignes de base Plaid et All-Aatom. Les échantillons à carreaux ont une meilleure diversité et capture le modèle bêta-brin qui a été plus difficile à apprendre pour les modèles génératifs de protéines.
Remerciements
Merci à Nathan Frey pour ses commentaires détaillés sur cet article et aux co-auteurs de Bair, Genentech, Microsoft Research et de l’Université de New York: Wilson Yan, Sarah A. Robinson, Simon Kelow, Kevin K. Yang, Vladimir Gligorijevic, Kyunghyun Cho, Richard Bonneau, Pieter Abbeel et Nathan C. Frey.
Source link